

Background: KRT-232 is a potent, selective, orally available, targeted inhibitor of human MDM2 homolog interactions with tumor suppressor protein 53 (p53). KRT-232 is under development for treatment of myeloproliferative neoplasms, acute myeloid leukemia and Merkel cell carcinoma. KRT-232 is a highly permeable 568.6 g/mol, monoprotic carboxylic acid (pKa 4.35); elimination involves primarily biliary-fecal excretion of an acyl glucuronide metabolite (M1) and enteric glucuronide hydrolysis with enterohepatic recirculation of parent drug. M1 is a 744.7 g/mol monoprotic acid with pKa 3.32 and 1/5th the MDM2 inhibitory potency of KRT-232. Plasma protein binding (fup) of KRT-232 and M1 were ~0.026 and ~0.006, respectively. Static model drug-drug interaction (DDI) calculations (FDA-CDER. 2020) indicated a potential minor DDI with substrates of cytochrome P450s (CYP) CYP3A4 and CYP2C8, UGT1A1, P-glycoprotein 1 (P-gp) and OATP1B1/3. Here, a 'fit-for-purpose' PBPK model for KRT-232 was developed using physicochemical, preclinical (in vitroandin vivo) and clinical pharmacokinetic (PK) data (KartosData on file; Ye et al.Xenobiotica. 2015; Gluck et al.Invest New Drugs. 2020 [Study A]).
Methods:Simcyp Simulator V17 (release 1; 27/11/2017; 17.0.90.0) was used.
In vitro kinetic parameters:In vitroKi for competitive inhibition of CYP2C8 was 3.8 and 0.17 µM for KRT-232 and M1, respectively.In vitroKI and kinact for mechanism-based inhibition of CYP3A4 by KRT-232 were 28 µM and 4.14 h-1, respectively. Estimatedin vitroKi values for KRT-232-mediated inhibition of UGT1A1, P-gp, OATP1B1 and OATP1B3, were 1.9, 14.8, 4.9 and 6.9 µM. For M1-mediated inhibition of UGT1A1, OATP1B1 and OATP1B3, values were 2.3, 0.393 and 0.606 µM.
Model development and verification:KRT-232 absorption was described by a first-order process; fraction absorbed was assumed to be 100% and the absorption rate constant was manually optimized using observed mean plasma concentration data from Part 1 of Study A. KRT-232 clearance was based onin vitroformation clearance of M1 in human liver microsomes, scaled up 4-fold based on observed oral clearance of KRT-232 (Wood et al.Drug Metab Dispos. 2017). KRT-232 distribution volume was described by a minimal PBPK model. KRT-232 and M1 DDI parameters were based onin vitrodata. M1 clearance was manually optimized using mean observed data from Study A (Part 1, 240 mg dose), and distribution volume was predicted by the Simulator using Method 1 and a full PBPK model. PBPK simulated data were compared with patient PK data collected during Part 2 of Study A.
Model application:The model predicted potential DDI after steady state (SS) 240 mg once daily KRT-232 administration with substrates of CYP3A4, CYP2C8, UGT1A1, P-gp and OATP1B1/3. DDI simulations included 100 virtual subjects assigned to 10 virtual trials (N=10 each). Population estimates of geometric mean and median Cmax and AUC0-∞ ratios were generated. A "worst case" sensitivity analysis was performed by using a 10-fold increase inin vitroCYP3A4 interaction potency (KI, EC50) relative to the measuredin vitrovalue.
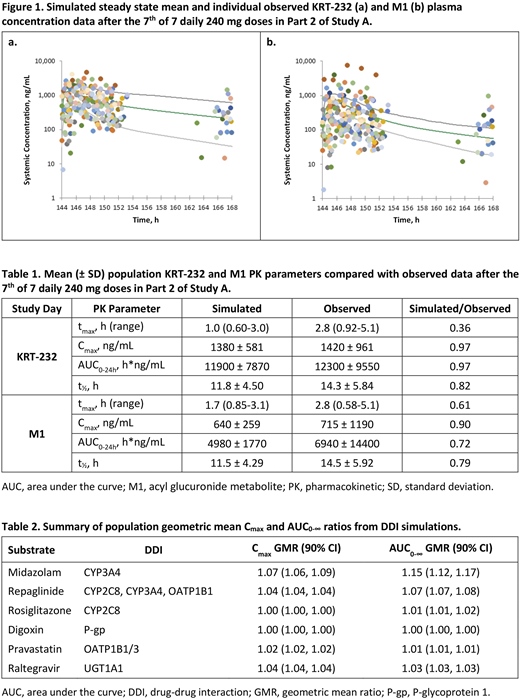
Results:Figure 1shows simulated SS mean and individual observed KRT-232 and M1 concentration data from Part 2 of Study A.
SS KRT-232 Cmax, AUC0-24h and t1/2 were predicted within 0.80-1.25-fold of observed PK data and tmax was under-predicted (0.36-fold) by the model (Table 1). M1 Cmax and AUC0-24h were within 0.80-1.25-fold of observed PK data on Day 1 (not shown). M1 Cmax on Day 7 was within 0.80-1.25-fold of observed PK data and SS AUC0-24h, t1/2, and tmax were underpredicted (0.72, 0.79 and 0.61-fold) relative to observed PK data.
In all simulated DDIs, no clinically significant interaction was predicted for SS 240 mg doses of KRT-232 since predicted Cmax and AUC0-∞ geometric mean ratio (GMR) changes were less than 1.25-fold (Table 2). The CYP3A4 DDI simulation gave a population GMR (90% CI) for Cmax and AUC0-∞ of 1.07 (1.06, 1.09) and 1.15 (1.12, 1.17), respectively. In a sensitivity check, predicted midazolam Cmax and AUC0-∞ after a 10-fold reduction ofin vitroKI and EC50 were 1.44-fold and 2.18-fold, respectively. Although unlikely, a weak CYP3A4 DDI could not be definitively excluded.
Conclusions: Mean plasma exposure of KRT-232 and M1 and inter-individual variability were adequately described by the PBPK model. No clinically significant DDI were predicted based on Cmax and AUC0-∞ GMR changes of less than 1.25-fold.
Templeton:Certara UK, LTD.:Current Employment.Podoll:IV/PO, LLC:Consultancy.Krejsa:Kartos Therapeutics:Current Employment;AstraZeneca:Current equity holder in publicly-traded company;Seattle Genetics:Current equity holder in publicly-traded company;Acerta Pharma:Current equity holder in private company.Slatter:Kartos Therapeutics:Current Employment;AstraZeneca:Current equity holder in publicly-traded company;Amgen:Divested equity in a private or publicly-traded company in the past 24 months.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal